
Computational Biology / Molecular Docking / Drug Screening
Closing the Gap
2026 • Computational Biology Research Project
A computational screening project focused on identifying which clinically relevant antibiotics showed the strongest predicted binding affinity against a Burkholderia cenocepacia protein target. The project used molecular docking to compare meropenem, ceftazidime, and minocycline as a first-pass method for reducing trial-and-error treatment selection in cystic fibrosis-related infections.
Status
Proof of Concept
Target
IspF Enzyme
Best Ligand
Minocycline
Role
Coding + Docking Workflow
Project Media
These visuals summarize the most important parts of the project: the docking workflow, the team method, and the final predicted binding affinity ranking.
Proof-of-Concept Result
Final ranking of the three antibiotic ligands by best predicted AutoDock Vina affinity in the selected active-site docking box.
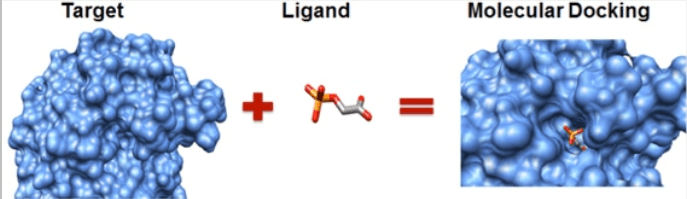
Molecular Docking Workflow
Conceptual workflow showing how a protein target and ligand are combined computationally to predict a molecular docking pose.

Team Capability + Methods
Project workflow summary covering team structure, responsibilities, docking tools, and why the scope was realistic for the course.
Project Motivation
Cystic fibrosis patients are especially vulnerable to lung infections because thick mucus buildup can make it harder to clear bacteria from the respiratory system. Burkholderia cenocepacia is a major concern because it can be drug-resistant, difficult to treat, and dangerous for patients who already have limited lung function.
Current treatment decisions can rely heavily on trial-and-error antibiotic combinations. Our project explored whether molecular docking could act as an early screening tool to compare antibiotic candidates before investing time and money into experimental testing.
Computational Workflow
The project followed a molecular docking workflow built from our course lab, then adapted it to compare three antibiotic ligands against the selected bacterial receptor.
Step 1
Protein Target
The receptor was selected as the IspF enzyme, a bacterial protein connected to the MEP pathway and bacterial survival.
Step 2
Ligand Selection
Meropenem, ceftazidime, and minocycline were chosen because they are clinically relevant antibiotics connected to drug-resistant bacterial infection research.
Step 3
File Preparation
Protein structures were cleaned and prepared with PDBFixer, while ligand structures from PubChem were converted into docking-ready formats using Meeko.
Step 4
Docking Simulation
AutoDock Vina was used to dock each antibiotic into a fixed active-site region based around the native C5P ligand location.
Step 5
Result Comparison
The team compared binding affinity values and ligand poses to identify which antibiotic showed the strongest predicted interaction with the receptor.
Antibiotic Candidates
We compared three antibiotic ligands with different biological mechanisms and clinical relevance to Burkholderia cepacia complex infection research.
Minocycline
A tetracycline antibiotic that interferes with bacterial protein production by interacting with the 30S ribosomal subunit.
Ranked first in the selected docking box with the strongest predicted Vina affinity.
Ceftazidime
A cephalosporin antibiotic connected to bacterial cell wall inhibition and commonly tested against Burkholderia cepacia complex isolates.
Ranked second and produced a strong negative binding score in the selected active-site box.
Meropenem
A carbapenem antibiotic used for serious multidrug-resistant Gram-negative bacterial infections and cell wall synthesis inhibition.
Ranked third in the selected box, while still showing favorable predicted binding.
Docking Visualizations
After running the docking simulations, we used Mol* to inspect the predicted ligand poses inside the selected receptor region.
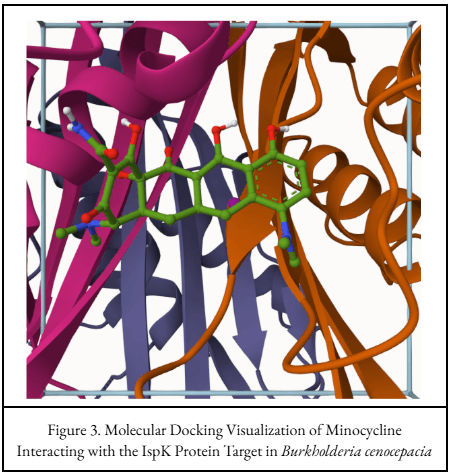
Minocycline Docking Pose
Mol* visualization of minocycline interacting with the selected Burkholderia cenocepacia protein target region.

Ceftazidime Docking Pose
Mol* visualization of ceftazidime bound within the selected docking region of the receptor.
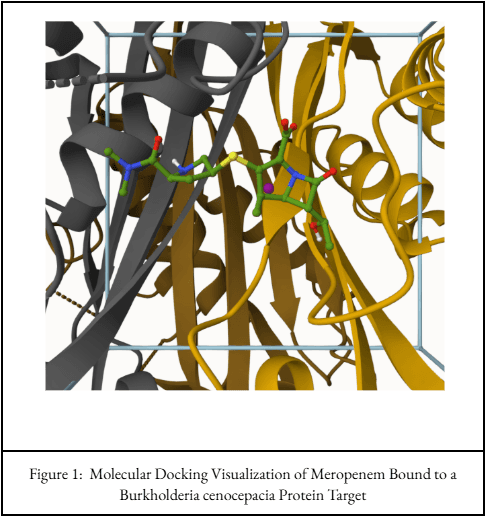
Meropenem Docking Pose
Mol* visualization of meropenem docked into the selected receptor region for comparison against the other antibiotics.
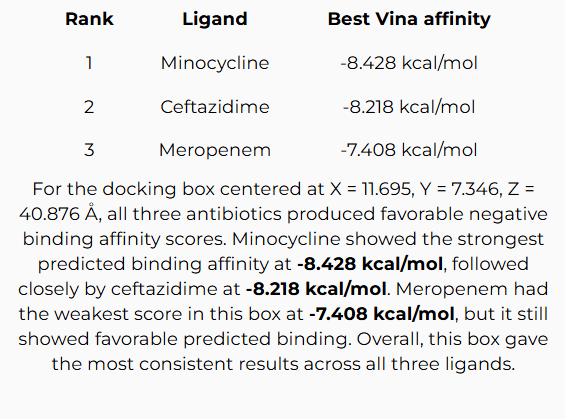
Proof-of-Concept Result
For the docking box centered at X = 11.695, Y = 7.346, Z = 40.876 Å with a 25 × 25 × 25 Å box size, all three antibiotics produced favorable negative binding affinity scores.
Rank 1
Minocycline
-8.428 kcal/mol
Strongest predicted binding affinity in the selected active-site docking box.
Rank 2
Ceftazidime
-8.218 kcal/mol
Second strongest predicted binding affinity, close behind minocycline.
Rank 3
Meropenem
-7.408 kcal/mol
Weakest of the three in this selected box, but still showed favorable negative binding.
Minocycline showed the strongest predicted binding affinity at -8.428 kcal/mol, followed closely by ceftazidime at -8.218 kcal/mol. Meropenem ranked third at -7.408 kcal/mol, but still showed favorable predicted binding. Overall, this docking box gave the most consistent comparison across all three ligands.
Technical Highlights
Used molecular docking as a first-pass computational screening method to compare antibiotic candidates against a Burkholderia cenocepacia protein target.
Selected the IspF enzyme as the receptor because it is involved in the bacterial methylerythritol phosphate pathway, which supports essential isoprenoid production.
Prepared protein and ligand files using a workflow built from PDB structures, PDBFixer, PubChem ligand data, Meeko conversion, and AutoDock Vina docking.
Compared meropenem, ceftazidime, and minocycline using predicted Vina binding affinity and docking pose geometry.
Evaluated two docking box locations and selected the active-site box that produced the most consistent negative binding scores across all three antibiotics.
Used Mol* to visualize ligand poses, receptor geometry, docking boxes, and predicted antibiotic-protein interactions.
Tools / Software / Methods
This project was built from a structured molecular docking lab workflow and expanded into a team-based antibiotic screening proof of concept.
Limitations
The project was useful as a computational screen, but the results should be understood as predictions rather than final medical conclusions.
The docking results are computational predictions and do not prove biological effectiveness on their own.
The receptor was treated as rigid, meaning the protein structure did not flex or change shape during the simulation.
Real proteins are flexible, so flexible docking or molecular dynamics would be needed for a higher-fidelity model.
Docking box placement strongly affects predicted binding, so selecting a realistic active site was a major part of the project.
Wet-lab testing would be required to confirm whether the predicted interactions matter biologically.
Mission Log
Closing the Gap was a computational biology project focused on using molecular docking to compare antibiotic candidates for drug-resistant Burkholderia cenocepacia infection in cystic fibrosis patients. The problem was meaningful because these infections are difficult to treat and current treatment selection can rely heavily on trial-and-error antibiotic combinations.
Our team approached the project as a first-pass screening problem. Instead of trying to prove that a drug works biologically, we wanted to use simulation to identify which antibiotics were worth prioritizing for future testing. We selected the IspF enzyme as the protein target because it is connected to the bacterial MEP pathway, which is important for producing isoprenoids needed for essential bacterial function.
My main role was working through the coding and terminal-based docking workflow. I helped prepare and run simulations, manage files in VS Code, swap the correct ligands and docking locations, and collect the binding affinity outputs for comparison. The process was not extremely difficult because we had a lab workflow to build from, but it still required careful file organization and attention to detail.
One of the main challenges was making sure the correct drug, receptor, and docking box were being used for each run. Since we tested more than one docking location, it was easy to accidentally run a simulation for the wrong site or with the wrong ligand file. After working through that confusion, I became more comfortable reading terminal commands, organizing computational files, and checking that each docking run matched the intended setup.
The project was also a solid team experience. We divided work across antibiotic research, ligand and receptor preparation, docking simulations, Mol* visualization, and results analysis. I enjoyed working with the team because everyone contributed, stayed involved, and helped keep the project organized without turning it into something unnecessarily stressful.
Final Result
The final result was a proof-of-concept computational screening package with docking visualizations, binding affinity rankings, a written report, and a presentation. In the selected active-site docking box, minocycline produced the strongest predicted binding affinity, followed by ceftazidime and meropenem.
The most important takeaway was not that the simulation proved a treatment, but that molecular docking can help narrow down which antibiotics may be worth investigating further. As a first-pass research tool, this kind of workflow can reduce guesswork, guide future wet-lab testing, and connect computational engineering methods to real biomedical problems.
What I Learned
This project helped me understand how computational biology tools can be used to study real biomedical problems. I learned how molecules are prepared for fixed molecular docking, how docking boxes affect results, and how binding affinity scores can be used to compare ligand candidates.
It also made me more confident working in VS Code with multiple files, terminal commands, receptor files, ligand files, and output data. More broadly, it gave me practice working with a team, listening to different ideas, and turning a structured lab workflow into a complete research-style project.